As of April 3, 2022, coronavirus disease 2019 (COVID-19) has caused almost 6.2 million deaths worldwide. While in many developed nations, life is beginning to return to normal, nations that did not obtain sufficient vaccine doses are still struggling to contain the COVID-19 pandemic.
In a recent study posted to the medRxiv* preprint server, researchers conduct a massive exome-wide genome study to explore whether there are any connections between rare gene variants/deleterious mutations and disease outcomes following infection with the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
Study: Exome-wide association study to identify rare variants influencing COVID-19 outcomes: Results from the Host Genetics Initiative. Image Credit: angellodeco / Shutterstock.com
About the study
Over 28,000 individuals infected with SARS-CoV-2 and an additional 596,000 controls from 21 cohorts in 12 different countries were included in the final analysis. These individuals were of European, African, Admixed America, Middle Eastern, as well as South and East Asian ancestries. The mean age of participants was 55.7, and each group had an even distribution of the sexes.
Three different outcome phenotypes were examined. These included individuals with severe COVID-19, which included individuals who died or required invasive respiratory support such as mechanical ventilation or hospitalization.
The second outcome included any individual with COVID-19 who died or required hospitalization and susceptibility to infection. Finally, the third phenotype included any individual infected with SARS-CoV-2. For all three phenotypes, controls were individuals who were not classified as cases.
As rare deleterious variants can have large effect sizes, burden tests were devised to increase statistical power to test associations between rare variants and medically relevant outcomes. These tests collapse each variant into larger sets or variations, following which association can be tested between these groups and each outcome.
Deleterious variants in each gene were collapsed and individuals were given a score for each gene. Whereas a score of zero was given if the individual carried no deleterious variant, a score of one was provided if they carried at least one non-homozygous deleterious variant and a score of two was given if they carry at least one homozygous deleterious variant.
Deleterious variants were chosen using different masks including predicted loss of function (pLoF), predicted loss of function and indels of moderate consequence and missense variables (coding5), and a final mask that uses all variants in coding5 and adds extra missense variables (coding1). Analyses were performed for variants with minor allele frequency (MAF) between less than 1% and less than 0.1%.
The resulting scores for each mask were then regressed on each of the three phenotypes using logistic regression and controlled for age, sex, interactions between these factors, and variant genetic principal components. Analyses were performed separately for each cohort and ancestry using Firth regression, followed by a meta-analysis using a fixed effect inverse-variance weighted model.
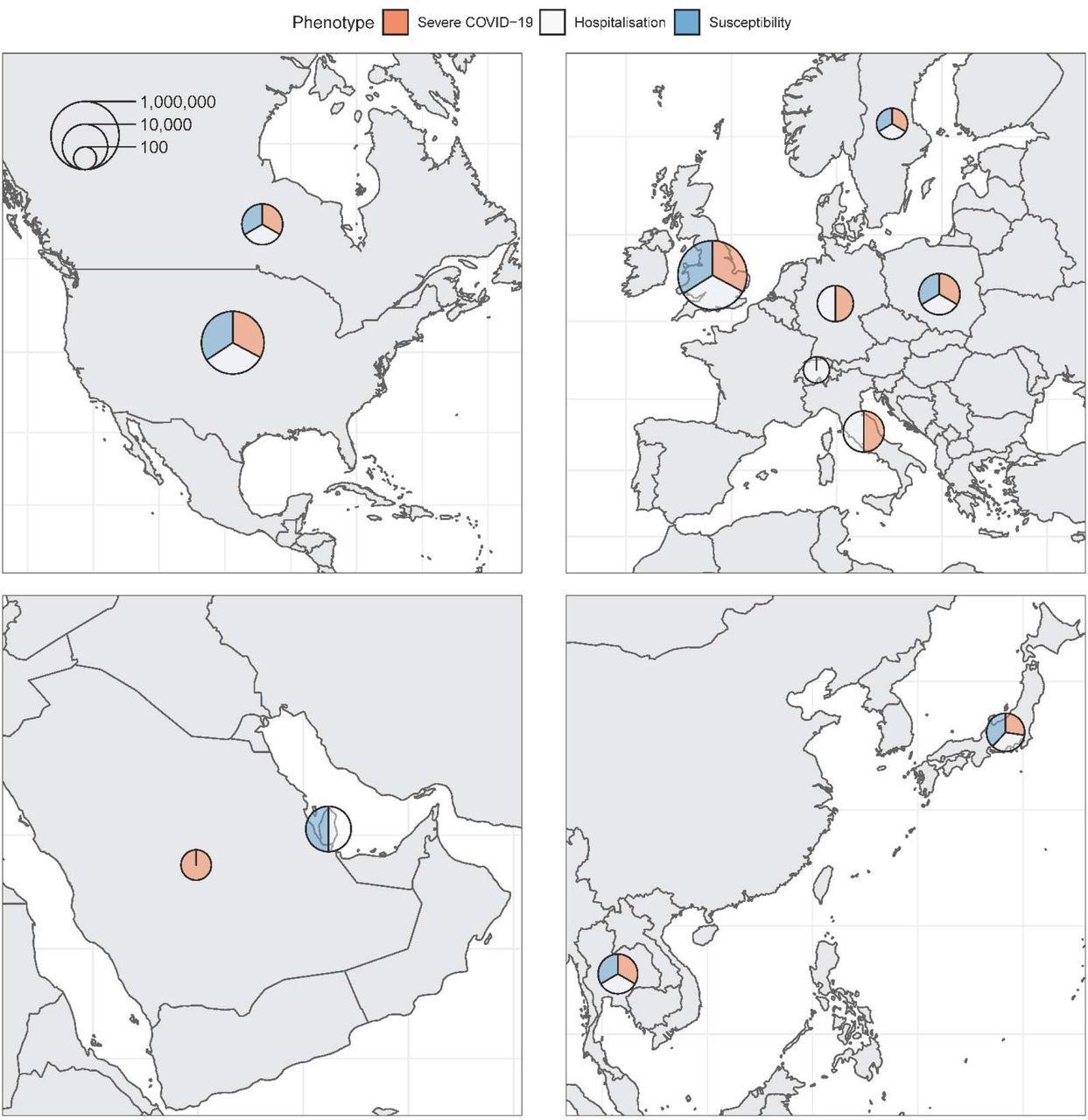
Maps of countries contributing data to the consortium. Sample sizes (cases and controls) for each phenotype were added and represented on the logarithmic scale by each circle. Relative contribution to each phenotype is represented by the three colors.
Study findings
Three genes were found to be associated with one of the COVID-19 phenotypes in at least one mask in the meta-analyses. Carrying either a pLoF or missense variant in the toll-like receptor (TLR7) gene was associated with a 5.3-fold increase in the odds of severe COVID-19. This finding was unsurprising, given that TLR7 plays an important role in viral immunity and upregulates the interferon pathways.
Analysis of TLR7 in other masks did not reach the statistical significance threshold. However, pLoFs in microtubule affinity regulating kinase 1 (MARK1) were also associated a 23.9 fold increased risk of severe COVID-19, as well as a 12.3-fold increase in the odds of hospitalization due to COIVD-19.
Although these mutations were not common, the signal was consistent across the three largest cohorts. MARK1 has previously been shown to interact with SARS-CoV-2 during infection.
The researchers could not replicate previously observed associations of genes in the interferon pathway with COVID-19 outcomes, even when more forgiving significance thresholds such as p=0.05 were used.
Testing for rare variant associations between genome-wide association study (GWAS) candidate genes showed an increased burden of pLoF or missense mutations in the ABO gene amongst those susceptible to SARS-CoV-2 infection. Often, those with deleterious variants in this gene show A or B blood groups, which previous studies have shown are more likely to be present in susceptible individuals.
Conclusions
The current study helped to confirm the effect of deleterious variants at TLR7 on individuals’ susceptibility to COVID-19, thus providing stronger statistical evidence than previous studies. In addition, the MARK1 gene findings uncover the potential role for cellular microtubule disruption during COVID-19, as well as provide evidence that contrasts prior findings on the impact of COVID-19 on the interferon pathway.
Taken together, the current study can help inform future genome and exome studies into SARS-CoV-2 infection and potentially assist in the identification of new treatment targets.
*Important notice
medRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- Butler-Laporte, G., Povysil, G., Kosmicki, J., et al. (2022). Exome-wide association study to identify rare variants influencing COVID-19 outcomes: Results from the Host Genetics Initiative. medRxiv. doi:10.1101/2022.03.28.22273040. https://www.medrxiv.org/content/10.1101/2022.03.28.22273040v2.
Posted in: Genomics | Medical Science News | Medical Research News | Disease/Infection News
Tags: Allele, Blood, Coronavirus, Coronavirus Disease COVID-19, Frequency, Gene, Genes, Genetic, Genetics, Genome, Genomic, immunity, Interferon, Kinase, Pandemic, Phenotype, Receptor, Respiratory, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Syndrome, Vaccine, Whole Genome Sequencing

Written by
Sam Hancock
Sam completed his MSci in Genetics at the University of Nottingham in 2019, fuelled initially by an interest in genetic ageing. As part of his degree, he also investigated the role of rnh genes in originless replication in archaea.
Source: Read Full Article