
Cystic fibrosis transmembrane conductance regulator (CFTR) modulators were a breakthrough for cystic fibrosis, improving the movement of chloride and water and moistening mucus secretions. But these drugs are expensive, don’t work in all patients with cystic fibrosis, and have side effects and interactions with other drugs. People who do respond to CFTR modulators must take them for a lifetime.
Researcher Ruby Wang, MD, and Benjamin Raby, MD, MPH, chief of the Division of Pulmonary Medicine at Boston Children’s Hospital, envision an alternative approach: cell therapy or gene therapy targeting a cell type that’s only recently been discovered.
Eyeing ionocytes
In 2018, two studies published in Nature rocked the cystic fibrosis scientific community. They found that the CFTR gene mutation primarily affects ionocytes—previously unknown cells that make up just 1% of the airway’s cells. Surprisingly, more than 90% of the CFTR protein was being made by these rare cells.
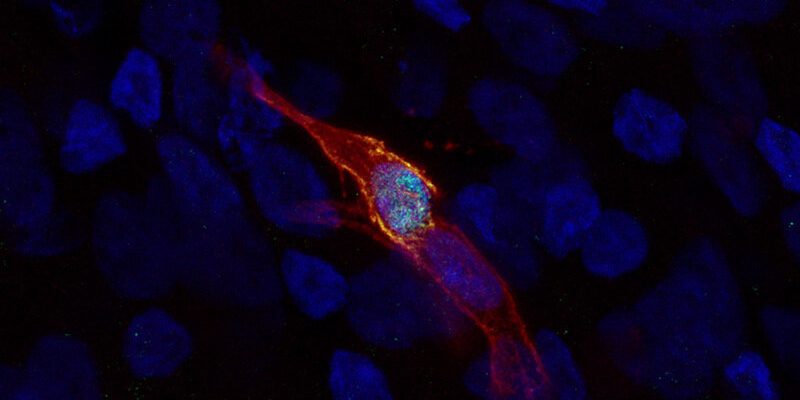
Now, in the American Journal of Respiratory and Critical Care Medicine, Wang and colleagues at Boston University report creating ionocytes from patients for the first time using stem cell technology. Collaborators at Boston Children’s include Thorsten Schlaeger, Ph.D., and Yang Tang from the Stem Cell Research Program and Stuart Rollins, MD, and Chantelle Simone-Roach in Pulmonary Medicine.
The accomplishment means that ionocytes can now be studied in a dish to understand their biology—and their possible use as a treatment vehicle.
“Maybe we could correct patients’ ionocytes and put them back in the lungs,” Wang says. “This is why it’s exciting.”
Overturning dogma in CF
The idea that ionocytes could play a key role in cystic fibrosis initially sparked intense skepticism. How could such an uncommon cell help keep the lungs clear? The airway’s ciliated cells, which propel mucus along the airways, seemed like the logical cell to target.
“When I was in medical school, the gospel was that the CFTR mutation was in the ciliated cells,” says Wang. “We didn’t believe in ionocytes at the time, and didn’t expect to make them.”
But to their surprise, they did. Starting with patients’ blood cells, Wang and her colleagues created induced pluripotent stem cells. They then directed the stem cells to differentiate in a stepwise fashion, first generating airway basal-like cells (iBCs) through a previously published protocol. Unexpectedly, modifying the protocol and stimulating the iBCs yielded ionocytes with high levels of the CFTR protein.
In a dish, the ionocytes have long extensions a bit like neurons. That has led the researchers to speculate that they might be talking to and influencing other cells. Wang thinks it will take more time to tease out exactly what they do.
“Now we need to find out ionocytes’ developmental origin and function,” says Wang. “That’s hard to study because they’re so rare.”
Wang’s work is part of a larger effort underway at Boston Children’s. With collaborators Carla Kim, Ph.D., Schlaeger, and George Daley, MD, Ph.D., of the Stem Cell Research Program, she and Raby have formed a Pulmonary Cellular Therapeutics Initiative to seek alternative therapies for lung diseases. Having the most relevant affected cells in hand, as well as a human-cell-based platform for studying them, cystic fibrosis could be the first test case.
More information:
Ruobing Wang et al, De Novo Generation of Pulmonary Ionocytes from Normal and Cystic Fibrosis Human Induced Pluripotent Stem Cells, American Journal of Respiratory and Critical Care Medicine (2023). DOI: 10.1164/rccm.202205-1010LE
Journal information:
Nature
,
American Journal of Respiratory and Critical Care Medicine
Source: Read Full Article