
The novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has undergone several rounds of mutations since it was first identified in Wuhan, China in December 2019. These mutations are attributed to genetic adaptations and modifications in different geographies, varying with population, ethnicity, and even gender.
 Study: Investigating the biological and technical origins of unknown bases in the S region of the SARS-CoV-2 Delta variant genome sequences. Image Credit: Naty.M / Shutterstock.com
Study: Investigating the biological and technical origins of unknown bases in the S region of the SARS-CoV-2 Delta variant genome sequences. Image Credit: Naty.M / Shutterstock.com
Sequencing the Delta variant
Sequencing SARS-CoV-2 and its mutated variants to understand their genetic composition is crucial. Particularly for public health purposes, this can help in designing vaccines more effectively and develop therapeutic cures for the disease in the future.
The latest mutation of SARS-CoV-2 was the Delta variant (B.1.617.2), which was first identified in India in July 2020 and subsequently spread to 115 countries. The Delta variant has been a prevalent topic of research in India, England, France, Germany, and Switzerland.
As per sequenced data from these countries, the emergence of new variants has been largely attributed to mutations in the spike (S) protein region of the viral genome, which is responsible for the binding and entry of SARS-CoV-2 to host cells and, as a result, its pathogenicity. Notably, the Delta variant includes the S:P681R and S:L452R mutations in the S region.
Despite using advanced sequencing methods to understand the genomic landscape of the SARS-CoV-2, the S region of the SARS-CoV-2 remains relatively under sequenced. In a recent study published on the preprint server medRxiv*, Swiss researchers have attempted to identify the possible technical drawbacks of current systems and decode the under sequenced region using improved techniques to understand the biological significance behind the Delta variant.
Current sequencing techniques
Whole-genome sequencing (WGS) is the method of choice for classifying genomic lineage and comparing viral isolates globally.
Two main processes ensure that the genomes are sequenced at high speed. These include amplicon tilling strategies, which allow the fast and reliable production of complete genome sequences based on sets of carefully chosen amplicons. These amplicons are products from polymerase chain reactions (PCR) with the genetic sequence of interest.
The second process is known as Next-Generation sequencing (NGS), which includes technologies that can sequence the resulting amplicons via short-read or long-read NGS technologies.
However, the amplicon-based methods have a major drawback. For example, these techniques may miss certain regions in the genome. This can lead to cases of unknown bases (hereby referred to as “N”) in the consensus sequences and may cause issues in identifying the lineage of viruses.
If there is a mutation in the target gene template used to design these primers, the chances of primer mismatch and the emergence of under sequence regions (USRs) increase.
Identifying and solving the problem
In the current study, the researchers used different sequencing methods to locate the USRs in Delta variant samples collected from the Institute for Infectious Diseases (IFIK) in Bern, Switzerland.
The researchers initially observed that the region surrounding the deletion 69-70 (nt positions) in the S region of SARS-CoV-2 Delta variant genome sequences was systematically under sequenced. They further observed unidentified bases from positions 21,357 to 22,346 of the genome sequence, consistently in all Delta variants (B.1.167.2, AY.1, AY.2, AY.3) from all major countries with prevalent Delta-variant infections (England, India, Germany, France, and the United States).
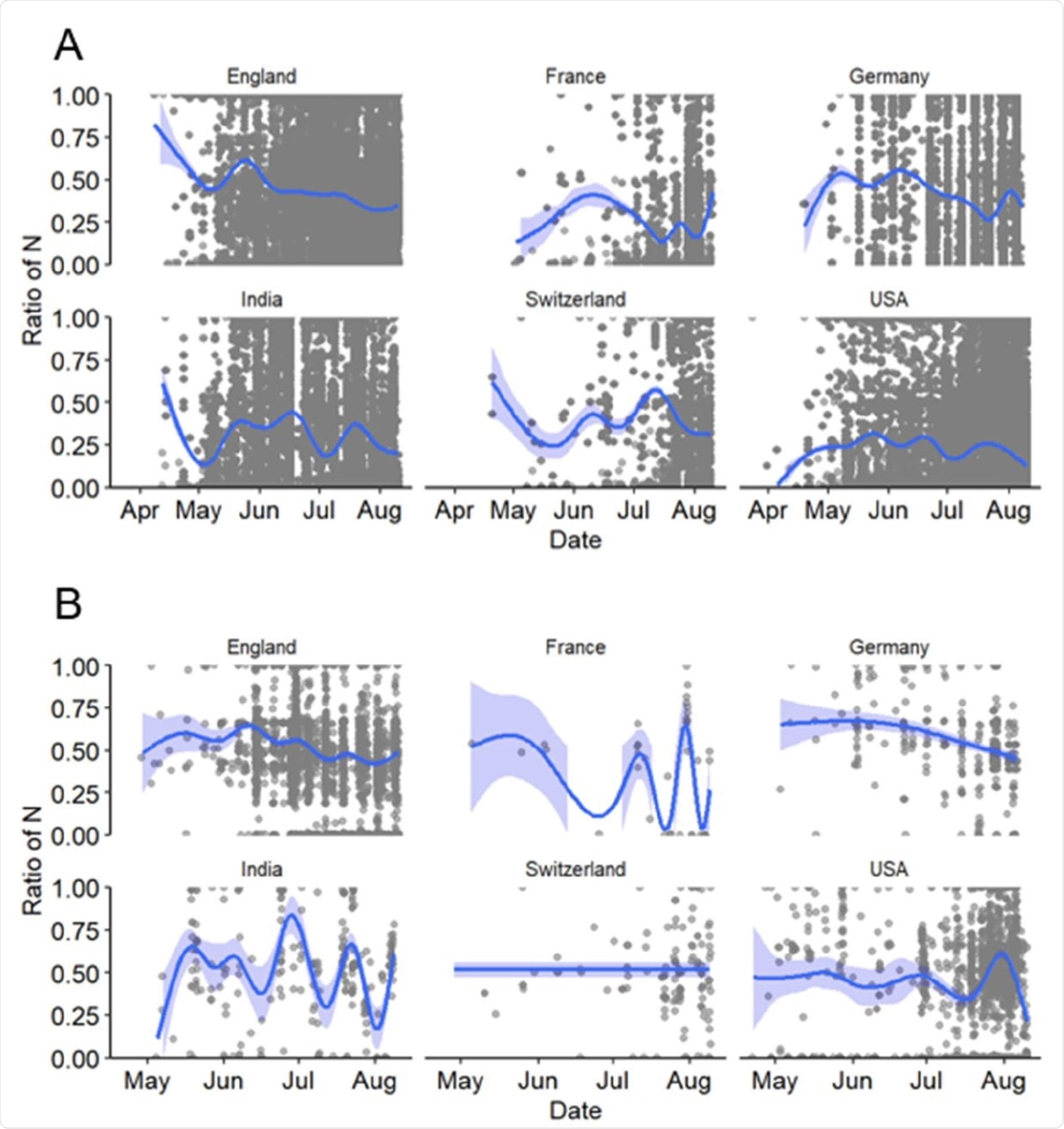 Ratio of number of Ns present in the ROI to the total number of Ns present in the entire genome of SARS-CoV-2 sequences for six countries in 2021. Considered here are A) all sequences with a total <5% of Ns and B) those with N-containing segments of length > 200 nt. The dark blue lines depict the best fitting lines of generalized additive models (GAM) with 95% confidence intervals (light blue areas).
Ratio of number of Ns present in the ROI to the total number of Ns present in the entire genome of SARS-CoV-2 sequences for six countries in 2021. Considered here are A) all sequences with a total <5% of Ns and B) those with N-containing segments of length > 200 nt. The dark blue lines depict the best fitting lines of generalized additive models (GAM) with 95% confidence intervals (light blue areas).
Primer mismatch was also identified for ARTIC v3 primers 72R and 73L to the sequences of the now predominant Delta variants. This mismatch was caused by deletion and mutational events.
The primer 72R, which is for binding to positions 22,013-22,038, displayed a truncated binding site due to a deletion between positions 22,029-22,034. Further, the primer 73L had a substitution (G21987A) for bindings between positions 21,961 to 21,990.
After demonstrating that the presence of this USR in the Region of Interest (ROI) of the viral genome was purely technical, the researchers designed alternative primers for primers 72R and 73L. This process specifically addressed the ARTIC v3 protocol, as it is the most widely used method for sequencing the coronavirus genome.
Implications
Since the primer-dependent sequencing methods are predominantly used for mapping the viral genomes, there are chances that mutations will alter the target sequence for these primers. The findings of this study indicated the possibility of such USRs existing in other regions of the genome of SARS-CoV-2 as well.
Overall, this study helped in understanding the basis of the appearance of SARS-CoV-2 mutants and how they can baffle the scientific community. This may lead to similar ‘waves’ of infection, as with the Delta variant.
Hence, it is of prime importance to regularly control for the presence of USRs and determine their prevalence in different geographical locations, amidst diverse populations and gene pools. Although it may be easier for countries working with smaller data pools to track the wet laboratory protocols used for SARS-CoV-2 genomic sequencing, it would be cumbersome for countries dealing with larger datasets.
Hence, data submitters should supply the necessary metadata on some of the key procedures involved in sequencing, like reverse transcription conditions, choice of primer sets, PCR amplification conditions, to name a few. Tracking these nuances in wet lab procedures will pave way for improved quality control in such cases and help in preventing transmission of more infectious strains in the future.
*Important notice
medRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- Borcard, L., Gempeler, S., Miani, M. A. T.,, et al. (2021). Investigating the biological and technical origins of unknown bases in the S region of the SARS-CoV-2 Delta variant genome sequences. medRxiv. doi:10.1101/2021.09.09.21262951. https://www.medrxiv.org/content/10.1101/2021.09.09.21262951v1
Posted in: Medical Science News | Medical Research News | Disease/Infection News
Tags: Bases, Coronavirus, Coronavirus Disease COVID-19, Gene, Genetic, Genome, Genomic, Genomic Sequencing, Infectious Diseases, Laboratory, Mutation, Polymerase, Protein, Research, Respiratory, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Syndrome, Transcription, Whole Genome Sequencing
Written by
Sreetama Dutt
Sreetama Dutt has completed her B.Tech. in Biotechnology from SRM University in Chennai, India and holds an M.Sc. in Medical Microbiology from the University of Manchester, UK. Initially decided upon building her career in laboratory-based research, medical writing and communications happened to catch her when she least expected it. Of course, nothing is a coincidence.
Source: Read Full Article