
 and MBNL2(–E6). Inclusion of Exon 6 results in a predicted nuclear localization signal. The cells were stained using DAPI (blue) and anti-FLAG (green) and anti-Myc (red) antibodies. Credit: Nucleic Acids Research (2023). DOI: 10.1093/nar/gkac1219")
Living organisms have a knack for persisting in the face of challenges. For instance, when genes malfunction, organisms may be able to compensate by activating redundant genes with similar functions, called paralogs.
One example of such compensation are two genes of the muscleblind-like (MBNL) family of RNA-binding proteins that lose their function in Myotonic Dystrophy Type 1, the most common cause of adult-onset muscular dystrophy. Loss of MBNL1 increases the levels of its paralog MBNL2 in tissues where protein expression is low, allowing MBNL2 to functionally compensate for MBNL1 loss. In animal models, loss of one paralog results in no or only minor functional consequences, whereas loss of both paralogs leads to a severe condition and lethality.
A paper on this topic is published in the journal Nucleic Acids Research.
“Although this form of compensation for loss of gene function has been widely observed, the underlying molecular mechanisms and roles in preventing or modifying disease severity are not well understood,” said first author Dr. Larissa Nitschke, a postdoctoral associate of pathology and immunology in Dr. Thomas A. Cooper’s lab at Baylor College of Medicine. “In this study, we discovered how loss of MBNL1 leads to the compensatory increase of MBNL2 protein.”
MBNL proteins are deeply involved in RNA processing, including alternative splicing, a process that allows cells to make many different proteins from a limited number of genes.
“To make proteins, genes in the DNA are transcribed into RNA, which is then translated into protein. Before RNA is translated into protein, it is processed and the fragments spliced in a certain way,” said Cooper, R. Clarence and Irene H. Fulbright Chair and S. Donald Greenberg Chair in Pathology. He also is the corresponding author of the work. “In almost all genes, the RNA is spliced in more than one way. That is alternative splicing; it allows one gene to make many different proteins. It would be like putting together different bracelets by combining a limited number of beads in unique ways.”
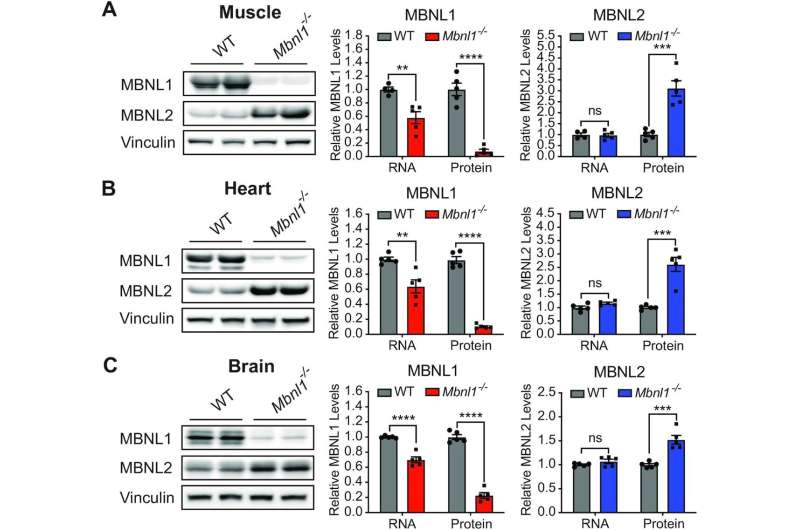
Nitschke, Cooper and their colleagues have found that loss of MBNL1 leads to the formation of MBNL2 with two new segments spliced to it, called exon 6 and exon 9.
“The inclusion of these exons gives MBNL2 new properties that enable it to compensate for the lack of MBNL1 function,” Nitschke said. “Inclusion of exon 6 promotes a more efficient transit of MBNL2 into the nucleus of the cell where this protein conducts its function. Adding exon 9 makes the protein less susceptible to degradation and more stable, which allows it to effectively compensate for MBNL1 loss of function.”
“Our findings could also provide an explanation for the high variability of the disease in terms of its features,” said Cooper, a member of the Dan L Duncan Comprehensive Cancer Center at Baylor. “Myotonic Dystrophy Type 1 can be very severe or mild, even in the same family. Individual differences in the ability to conduct these compensatory measures may explain the differences in severity.”
“The discovery of the compensatory mechanism also supports further studies to explore, if similar to the splicing-enhancing drugs in Spinal Muscular Atrophy, the mechanism could be leveraged for therapeutic purposes in Myotonic Dystrophy Type 1,” Nitschke said.
More information:
Larissa Nitschke et al, Alternative splicing mediates the compensatory upregulation of MBNL2 upon MBNL1 loss-of-function, Nucleic Acids Research (2023). DOI: 10.1093/nar/gkac1219
Journal information:
Nucleic Acids Research
Source: Read Full Article